What Is the Biological Definition of a Autosomal Dominant Disorder
Introduction
Polycystic kidney disease (PKD) is a heterogeneous group of monogenic disorders characterized by the bilateral formation and progressive expansion of renal cyst that lead to end stage renal disease (ESRD) (1). Several Mendelian diseases including autosomal dominant polycystic kidney disease (ADPKD), autosomal recessive polycystic kidney disease (ARPKD), and atypical forms of PKD can be grouped under this pathological entity.
Autosomal dominant polycystic kidney disease is the most common inherited kidney disease affecting ~1/400–1/1,000 individuals (2). The hallmark characteristic of ADPKD is the progressive development and expansion of cysts in the kidney leading to ESRD. It can be associated with several extrarenal manifestations including hypertension, symptomatic extrarenal cysts, and subarachnoid hemorrhage from intracranial aneurysms (3–5). The vast majority of the patients develop the disease between the ages of 20–40 s, but there have been sporadic cases that range in onset from late to childhood ("early onset," before 15 years old) or even in utero ("very early onset") (6).
Genetics of the ADPKD
Autosomal dominant polycystic kidney disease is genetically heterogeneous and associated with mutations in PKD1 (responsible of ADPKD-Type I), PKD2 (-Type II), and GANAB. PKD1 is a complex gene mapping to chromosome 16 (16p13.3) (Figure 1A). Its genomic structure has a number of features that complicate its evaluation (7, 8): (a) it is highly GC-rich with a large number of CpG dinucleotides, (b) 70% of PKD1 is duplicated multiple times throughout chromosome 16 with high sequence fidelity (95% identity) (9), and (c) it contains a 2.5 kb polypyrimidine tract in intron 21 (the largest in the human genome) (10). In contrast to PKD1, the PKD2 gene is located on chromosome 4 (4q21) and has simpler features and structure (11) (Figure 1A). Approximately, 80–85% of ADPKD families were associated with PKD1 mutations, and 15–20% to PKD2 mutations in the literature (12). Recently, Porath and colleagues identified causal mutations in GANAB, a gene on chromosome 11q12.3 (Figure 1A), in ADPKD patients that are negative for PKD1 and PKD2 mutations. They report that GANAB accounts for ~0.3% of total ADPKD and it is associated with a milder manifestation of PKD and autosomal dominant polycystic liver disease (ADPLD) (5).
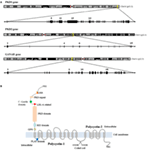
Figure 1. Chromosome localization and genomic structure of PKD1, PKD2, and GANAβ genes and structure of polycystin-1 (PC1) and polycystin-2 (PC2). (A) Schematic representation of chromosomes and genomic structure for the genes. (B) Representation of PKD1 and PKD2 protein products: PC1 and PC2. *GIIα (encoded by GANAβ) is not included because the tertiary structure of the protein is not available in the bibliography or protein date bases.
Diagnosis of ADPKD
The diagnosis of ADPKD is dependent on the stage of the disease. When the disease is fully established, the diagnosis is clinically based on patient's history and physical examination (13, 14). However, definitive diagnosis can be difficult due to other disorders having overlapping symptoms. Therefore, complementary approaches such as diagnostic imaging or genetic tests are necessary to confirm the diagnosis. Imaging techniques, including ultrasound, computed axial tomography, or nuclear magnetic resonance, allow for the detection of cysts in the kidney, liver, or pancreas (15). The magnetic resonance technique has proven to be more sensitive than ultrasound, allowing measurements of height-adjusted total kidney volume (htTKV) and better definition of the cysts without the use of contrast agents. However, these imaging tests are expensive (16) and are often not performed on a subset of the ADPKD population including those who are young individuals at risk or patients with atypical or de novo renal cystic disease (13) for whom complementary genetic tests is the method for definitive diagnosis. Direct DNA sequencing (DS) could offer a molecular diagnosis; however, the genetic analysis of the PKD1 (responsible for most ADPKD cases) is complicated. The 5′-region of the gene (exon 1–34) is replicated in at least six highly homologous copies on chromosome 16 (7, 9, 17). To date, direct sequencing based on a Long-Rage PCR strategy with specific primers has been the accepted strategy by the ADPKD community (18). Isolated gene by gene sequencing is laborious and expensive, and provides limited amount of information to provide a better diagnosis and prognosis for the patients. Moreover, it has been described that the main mutation responsible of the disease may interact with other PKD or ciliopathy loci modifying the phenotype and extending the genetic complexity of the disease (18–20). For this reason, ADPKD experts are highlighting the necessity to screen all cystic genes in a common strategy to allow for a more accurate diagnosis, including those genes responsible of atypical forms of PKD. Under this context, next-generation sequencing strategies followed by the validation of variants by DS have become the recommended methodology allowing for faster, more cost-effective, and more reliable genetic diagnosis of large ADPKD cohorts (21).
Genotype–Phenotype Correlations
It has been described that patients with mutations in PKD1 gene have larger kidneys and earlier onset (mean age at ESRD, 53.4 versus 72.7 years old, respectively) with lower eGFR and higher htTKV than PKD2 patients (22, 23). In addition, GANAB mutations seem to be associated with a mild renal phenotype, closer to a PKD2 than a PKD1 phenotype, revealing the importance of molecular diagnosis (5). Moreover, a strong correlation between the type of the mutation and the severity of the disease was observed, illustrating the importance of quantifying genetic heterogeneity in ADPKD. Truncating PKD1 mutations (frameshift, splicing, and nonsense) have a more severe disease prognosis with lower eGFR; however. the type of mutation does not correlate with htTKV (23, 24). Non-truncating PKD1 mutations (missense, inframe deletion/insertion) or mutations in PKD2 are associated with a milder form of the disease. In addition, males with truncating PKD1 are associated with larger kidneys and increased risk for ESRD, while women with truncating PKD1 have a more severe liver phenotype (23, 24). In addition, disease manifestation in ADPKD patients from the same family, or patients with the same mutation, can have varying severity and differential disease progression, which may be due to the presence of variation in a modifying gene. This phenomenon is known as genetic interaction and epistasis, and usually aggravates or attenuates the phenotype cause by the primary mutation (17, 25).
Based on genetic and clinical data, Cornec-Le Gall and colleagues (26) developed a robust prognostic model, the PROPKD score (with a range from 0 to 9), to predict survival in ADPKD patients. They described critical variables associated with ESRD including age of onset (median age reported to be 70.6 years for low risk, 56.9 years for intermediate risk, and 49 years for high risk) and a scoring system to predict disease progression: sex (being male 1 point), need for antihypertensive therapy before 35 years old (2 points), occurrence of the first urologic event before 35 years old (2 points), and genetic status (having PKD2 mutations: 0 points, non-truncating PKD1 mutation: 2 points and truncating PKD1 mutations: 4 points). Three risk categories were then defined to describe the putative risk for progression to ESRD: low risk (0–3 points), intermediate risk (4–6 points), and high risk (7–9 points) (26).
PKD Proteins: Structure and Function
PKD1 and PKD2 encode the proteins PC1/Polycystin-1 and PC2/Polycystin-2 or TRPP2, respectively. PC1 is a putative receptor for an unidentified ligand which contains a long extracellular N-terminal domain, 11 transmembrane domains and a short intracellular C-terminal domain (27). PC2/TRPP2 has similar characteristics to TRP channel, having six transmembrane segments, a pore loop domain (separating the first two transmembrane segments), and an N- and C-terminal domains (28) (Figure 1B). PC2 is a Ca2+-permeable non-selective ion cation channel and together with PC1 forms a receptor–channel complex implicated in the Ca2+ pathway called PC complex (29).
In contrast to PKD1 and PKD2, GANAB encodes the alpha subunit of glucosidase II (GIIα) which is the catalytic subunit of GII. GIIα together with the regulatory subunit of GII, GIIβ (also called hepatocystin) (5) form a functional holoenzyme in the endoplasmic reticulum (ER). This holoenzyme is implicated in the proper folding and translocation of glycoproteins into the ER, and its dysfunction has been reported to be associated with maturation and localization defects of PC1 (30).
Disease Mechanism
Two-Hit Model for ADPKD
The human kidney has approximately one million nephrons, and an ADPKD patient will develop around a 1,000 cysts (31). ADPKD disease progression is highly variable and depends directly from the nature of the mutated gene. The "Two-Hit Model," in which two different mutations affect proper genetic/cellular interactions, has been the proposed theory to explain the kidney phenotype observed in ADPKD patients. While an individual has inherited a germ line mutation ("first hit"), the development of cysts does not occur until another mutation (somatic mutations) in either PKD1 or PKD2 occurs ("second hit") (32, 33).
Localization of PKD Proteins: The Role of Primary Cilia
There have been a number of different localizations proposed for the PC1/PC2, including the ER, apical and basolateral cell membranes, or secreted exosomes (31). However, there is evidence supporting their presence in primary cilia based on their central role in ADPKD pathogenesis. Cilia are microtubule-based, non-motile organelles on the apical surface of the cells and play an essential role in cellular detection and regulation of external signals. Dysfunction of this organelle result in a group of disorders called the ciliopathies (34). Data from animal models (C. elegans, Drosophila, and Mus musculus) support the idea that defects in function or structure of primary cilia contribute to the pathomechanisms of PKD (35). PKD proteins such as PC1, PC2, and polyductin/FPC (encoded from the ARPKD gene, PKHD1) localized to the primary cilium (36–38). These PKD proteins interact with each other (17, 29, 36, 39, 40) and form a functional complex with common downstream signaling pathways (41). In addition, deleted in azoospermia interacting protein 1-like, the protein encoded from the recently identified ARPKD gene (DZIP1L), was reported to localize to the centrioles and basal bodies of cilia and are also associated with ciliary trafficking defects (19).
There has also been additional evidence to support the functional role of PKD proteins within the cilium. Urine flow has been linked to an increase in intracellular calcium (42), likely driven by the mechanical response of the primary cilium (43). The large extracellular domain of PC1 has been proposed to be the flow mechanosensor that opens the PC2-channel, allowing calcium entry leading to mechanotransduction activation (44). A different model proposes that the primary cilia's role in flow sensing is required for proper centrosomal localization that results in oriented cell division (OCD). In addition, defects in cilia drive the loss of planar cell polarity and consequently abnormal OCD (45); however, this model is controversial and remains unclear (46, 47). Several observations support the idea of the mechanosensory role of polycystins in the primary cilium (48, 49). PC2 directly interacts with KIF3A and KIF3B, two essential proteins for ciliary assembly and function (37, 50). In addition, PC2 is required for the flow-mediated increase of cytosolic Ca2+ (51, 52), and mechanical stimuli can induce proteolytic cleavage of the intracellular C-terminal domain of PC1 (53). Interestingly, there have been controversial results reporting that mechanosensation does not occur via Ca2+ signaling within cilia (54). In spite of this, there are some unanswered questions as while Delling and colleagues do not exclude the presence of others mechanosensitive elements in primary cilia (55) and the cilia seems to increase cytoplasmic Ca2+ concentration by diffusion (56).
A very interesting and unexpected finding by Ma and colleagues showed that loss of cilia results in a significant reduction of PKD severity (57). Authors reported that a simultaneous inactivation of polycystins and cilia assembly resulted in the reduction of the cystic phenotype associated with polycystins inactivation. These findings suggest that the polycystins modulate a pathway involved in the cilia signaling, but require intact cilia function (58).
Threshold or Dosage Model
Genetic background influences the phenotypic variability of ADPKD. As we previously mentioned, patients with mutations in PKD1 have worse prognosis than those with mutations in PKD2 (59), and those with truncating PKD1 mutations were associated with more severe polycystic renal pathology than those with non-truncating mutations (60). In addition, unaffected patients who carry a missense variant in PKD1 indicate that some alleles are incompletely dominant in the disease (61). Similarly, other studies suggest that incomplete, penetrant alleles can influences disease severity in ADPKD (62).
These data support that a threshold or dosage model could explain cystogenesis in ADPKD (63). According to this model, cyst initiation and cystic expansion depends on PKD gene dosage, starting when the level of functional PC falls below the cystogenic threshold (58, 63). Defects in that threshold may occur by a combination of one or more factors: the nature of germline mutation ("first-hit"), somatic mutations ("second hit"), modifier genes or environmental factors such as renal injury or inflammation (58, 64). Several studies support this: (1) García-González and colleagues reported genetic interaction between ADPKD and ARPKD genes in a common pathway (17), (2) it has been reported that ADPLD genes (Prkcsh and Sec63), ARPKD gene (Pkhd1) and ADPKD gene (Pkd1) interact with each other suggesting a central role of PC1 in cystogenesis (65), and (3) a developmental window for cystogenesis has been identified, suggesting that timing of secondary events may influence the severity of ADPKD (66).
A crucial step in the protein maturation of functional PCs is also related to the dosage model. Autoproteolytic cleavage of PC1 at the GPS domain, mediated by larger GAIN [G protein-coupled receptor (GPCR)-autoproteolysis inducing] domain which includes a GPCR proteolysis site (GPF) motif, is crucial for PC1 maturation (67, 68). Besse and colleagues have described that specific isolated-PLD proteins (encoded by SEC61β, ALG8, and GANAβ), from the ER protein biogenesis pathway, are directly related to PC1 biogenesis (30). Furthermore, PC1 maturation requires PC2 in a dose-dependent manner (69). It is also known that mature PC assembles at the PC complex-bearing vesicles in the Golgi before trafficking to the ciliary/plasmatic membrane (70) (Figure 2). In addition, Cai and colleagues described the effect of several mutations in Pkd1 and Pkd2 in the importance of PCs trafficking to cilia using in vitro and in vivo models, concluding that altered trafficking and dysfunctional maturation of PC complex underlie PKD pathology (71) These facts suggest a central role for PC1 in the cystogenesis process and in regulating the severity of ADPKD, ARPKD, and ADPLD (72).
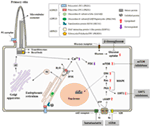
Figure 2. Diagram of the localization of the polycystic kidney disease (PKD) proteins, the pathways implicated in its pathogenesis, and putative therapeutic targets. The polycystin-1 (PC1) and polycystin-2 (PC2), associated with autosomal dominant polycystic kidney disease (ADPKD), and FPC and DIZP1L, associated with autosomal recessive polycystic kidney disease (ARPKD), are ciliary proteins and have functions in relation with the primary cilia. GANAβ [associated with ADPKD and autosomal dominant polycystic liver disease (ADPLD)], together with the classical genes of the ADPLD, PRKCSH, and Sec63, is localized in the endoplasmic reticulum (ER) and plays a role in the translocation and folding of the protein maturation. PC1 and PC2 form a receptor–channel complex in the cilium and is implicated in the Ca2+ pathway. PC2 also regulates intracellular calcium in the ER. PC mutations result in deregulation of Ca2+ leading an increase in cAMP and upregulation of the PKA and MAPK pathways. Abbreviations: RyR, ryanodine receptor; IP3R, IP3 receptor; PDE, phosphodiesterase; AC-VI, adenylyl cyclase 6; Gs and Gi, guanosine nucleotide-binding proteins; V2R, V2 receptor; cAMP, cyclic AMP; PKA, protein kinase A; MAPK, MAP kinases; SIRT1, sirtuin 1.
Signaling Pathways and Targeted Therapies in ADPKD
Several signaling pathways and transcription factors control the progression and development of cystogenesis (64, 73). Calcium signaling is one of the most studied pathways in the PKD field. PC2 is a calcium permeable non-selective cation channel that is abundantly expressed in the ER and interacts with others calcium channel proteins (51). It binds to the inositol 1,4,5-trisphosphate receptor (IP3R) regulating Ca2+ homeostasis and the activity of ryanodine receptors (74, 75). In contrast to PC2, PC1 accelerates the decay of the intracellular calcium response to ATP by increasing ER calcium uptake. All of this suggests a major role of polycystins in intracellular calcium hometostasis (76, 77). Cystic epithelial cells have an aberrant cross talk between intracellular calcium and cAMP signaling as elevated levels of cAMP stimulate cyst fluid secretion, enhancing protein kinase A activity (78–80). Furthermore, V2 receptor antagonists (Tolvaptan) ameliorate the progression of PKD by the inhibition of cAMP signaling pathway in both animal models (81, 82) and in clinical trials (83) (Figure 2). Importantly, adverse secondary effects could appear with Tolvaptan treatment such as polyuria, nocturia, and elevation of liver enzymes (84). Taking this into account, Tolvaptan is the first therapy approved for indication of ADPKD in several countries.
Alterations to other pathways have been reported to affect cystic volume or cystic progression, but to date; attempts to completely inhibit cystogenesis have been unsuccessful. The mTOR pathway is highly activated in cystic tissues independent of the PKD gene mutation (85). Preclinical trials with sirolimus and everolimus blocked cystic progression in a rodent model of ADPKD (86, 87). In addition, somatostatin analogs, as octreotide and lanreotide reduced hepatic and renal volume expansion in ADPKD (88–90). Treatment of PKD animal models, which have defective glucose metabolism associated with cystic expansion, with 2-deoxyglucose, an analog of glucose, also result in reduced cystic progression (91, 92) (Figure 2). Adding to the hunt for therapeutics, other alternate mechanisms also exist, such as sirtuin 1, microRNAs, and MCP1 which have been postulated as possible therapies for PKD (93–95) and recently, ongoing Phase-II and Phase-I clinical trials of a multi-kinase inhibitor, tesevatinib, are ongoing for ADPKD and children with ARPKD (96, 97).
Conclusion
In the last two decades, there have been significant contributions toward our understanding of the genetic, cellular, and functional role of PKD genes and proteins, as well as, the identification of a number of pathways implicated in the pathogenesis of the disease. Nevertheless, several questions remain unresolved and controversial in the PKD community. The complexity of this disease is reflected along all scientific levels, starting at the genetic level with critical refinement of the mutations, followed by the study of protein function and dosage to understand the spectrum of clinical manifestations in PKD, and finally the study of related pathways and modifier mechanisms that all should be taken into account for future clinical trials and personalized medicine.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank María Pardo and Perciliz Tan for their comments.
Funding
This work was supported by grants from the Instituto de Salud Carlos III FEDER funds ISCIII RETIC REDINREN RD12/0016, PI11/00690, PI15/001467, Sociedad Española de Nefrología, and Xunta de Galicia.
References
1. Lee K, Battini L, Gusella GL. Cilium, centrosome and cell cycle regulation in polycystic kidney disease. Biochim Biophys Acta (2001) 1812(10):1263–71. doi:10.1016/j.bbadis.2011.02.008
CrossRef Full Text | Google Scholar
2. Reiterová J, Štekrová J, Merta M, Kotlas J, Elišáková V, Lněnička P, et al. Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol (2013) 14:59. doi:10.1186/1471-2369-14-59
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol (2010) 6(4):197–206. doi:10.1038/nrneph.2010.18
CrossRef Full Text | Google Scholar
5. Porath B, Gainullon VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, et al. Mutations in GANAB, encoding glucosidase IIα subunit, cause autosomal dominant polycystic kidney disease and liver disease. Am J Hum Genet (2016) 98(6):1193–207. doi:10.1016/j.ajhg.2016.05.004
CrossRef Full Text | Google Scholar
7. Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet (1995) 10(2):151–60. doi:10.1038/ng0695-151
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Burn TC, Connors TD, Dackowski WR, Petry LR, Van Raay TJ, Millholland JM, et al. Analysis of the genomic sequence for the autosomal dominant polycystic kidney disease (PKD1) gene predicts the presence of a leucine-rich repeat. Hum Mol Genet (1995) 4(4):575–82. doi:10.1093/hmg/4.4.575
CrossRef Full Text | Google Scholar
9. Bogdanova N, Markoff A, Gerke V, McCluskey M, Horst J, Dworniczak B. Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics (2001) 74(3):333–41. doi:10.1006/geno.2001.6568
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Van Raay TJ, Burn TC, Connors TD, Petry LR, Germino GG, Klinger KW, et al. A 2.5kb polypyrimidine tract in the PKD1 gene contains at least 23 H-DNA-forming sequences. Microb Comp Genomics (1996) 1(4):317–27. doi:10.1089/mcg.1996.1.317
CrossRef Full Text | Google Scholar
12. Cornec-Le Gall E, Audrézet MP, Renaudineau E, Hourmant M, Charasse C, Michez E, et al. PKD2-related autosomal dominant polycystic kidney disease: prevalence, clinical presentation, mutation spectrum and prognosis. Am J Kidney Dis (2017) 6386(17):30537–30531. doi:10.1053/j.ajkd.2017.01.046
CrossRef Full Text | Google Scholar
14. Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol (2002) 13(9):2384–98. doi:10.1097/01.ASN.0000028643.17901.42
CrossRef Full Text | Google Scholar
15. Mosetti MA, Leonardou P, Motohara T, Kanematsu M, Armao D, Semelka RC. Autosomal dominant polycystic kidney disease: MR imaging evaluation using current techniques. J Magn Reson Imaging (2003) 18(2):210–5. doi:10.1002/jmri.10347
CrossRef Full Text | Google Scholar
16. Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol (2015) 26(1):160–72. doi:10.1681/ASN.2013101138
CrossRef Full Text | Google Scholar
17. Garcia-Gonzalez M, Menezes L, Piontek K, Kaimori J, Huso D, Watnick T, et al. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum Mol Genet (2007) 16(16):1940–50. doi:10.1093/hmg/ddm141
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Garcia-Gonzalez MA, Jones JG, Allen SK, Palatucci CM, Batish SD, Seltzer WK, et al. Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease. Mol Genet Metab (2007) 92(1–2):160–7. doi:10.1016/j.ymgme.2007.05.004
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Lu H, Rondón-Galeano M, Ott E, Kaeslin G, Kausalya P, Kramer C, et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet (2017) 49(7):1025–34. doi:10.1038/ng.3871
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Hartung E, Guay-Woodford L. DZIP1L defines a new functional zip code for autosomal recessive PKD. Nat Rev Nephrol (2017) 13(9):519–20. doi:10.1038/nrneph.2017.102
CrossRef Full Text | Google Scholar
21. Renkema KY, Stokman MF, Giles RH, Knoers NV. Next-generation sequencing for research and diagnostics in kidney disease. Nat Rev Nephrol (2014) 10(8):433–44. doi:10.1038/nrneph.2014.95
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Harris PC, Bae KT, Rossetti S, Torres VE, Grantham JJ, Chapman AB, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol (2006) 17(11):3013–9. doi:10.1681/ASN.2006080835
CrossRef Full Text | Google Scholar
23. Heyer CM, Sundsbak JL, Abebe KZ, Chapman AB, Torres VE, Grantham JJ, et al. Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol (2016) 27(9):2872–84. doi:10.1681/ASN.2015050583
CrossRef Full Text | Google Scholar
24. Hwang YH, Conklin J, Chan W, Roslin NM, Liu J, He N, et al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol (2016) 27(6):1861–8. doi:10.1681/ASN.2015060648
CrossRef Full Text | Google Scholar
25. Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol (2011) 22(11):2047–56. doi:10.1681/ASN.2010101080
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Cornec-Le Gall E, Audrézet MP, Rousseau A, Hourmant M, Renaudineau E, Charasse C, et al. The PROPKD Score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol (2016) 27(3):942–51. doi:10.1681/ASN.2015010016
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Hanaok K, Qian F, Boletta A, Bhunia A, Piontek K, Tsiokas L, et al. Co-assembly of polycystin-1 and -2 produces unique cation-permeable current. Nature (2000) 408(6815):990–4. doi:10.1038/35050128
CrossRef Full Text | Google Scholar
30. Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest (2017) 127(9):3558–3558. doi:10.1172/JCI96729
CrossRef Full Text | Google Scholar
31. Grantham J. Clinical practice: autosomal dominant polycystic kidney disease. N Engl J Med (2008) 359(14):1477–85. doi:10.1056/NEJMcp0804458
CrossRef Full Text | Google Scholar
32. Qian F, Watnick T, Onuchic L, Germino G. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell (1996) 87(6):979–87. doi:10.1016/S0092-8674(00)81793-6
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Watnick T, Torres VE, Gandolph MA, Qian F, Onuchic LF, Klinger KW, et al. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell (1998) 2(2):247–51. doi:10.1016/S1097-2765(00)80135-5
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Gerdes J, Davis E, Katsanis N. The vertebrate primary cilium in development, homeostasis and disease. Cell (2009) 137(1):32–45. doi:10.1016/j.cell.2009.03.023
CrossRef Full Text | Google Scholar
36. Yorder B, Hou X, Guay-Woodford L. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol (2002) 13(10):2508–16. doi:10.1097/01.ASN.0000029587.47950.25
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Wu Y, Dai X, Li Q, Chen C, Mai W, Hussain Z, et al. Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum Mol Genet (2006) 15(22):3280–92. doi:10.1093/hmg/ddl404
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Wang S, Zhang J, Nauli S, Li X, Starremans P, Luo Y, et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol (2007) 27(8):3241–52. doi:10.1128/MCB.00072-07
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Kim I, Fu Y, Hui K, Moeckel G, Mai W, Li C, et al. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am Soc Nephrol (2008) 19(3):455–68. doi:10.1681/ASN.2007070770
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Kaimori J, Germino G. ARPKD and ADPKD: first cousins or more distant relatives? J Am Soc Nephrol (2008) 19(3):416–23. doi:10.1681/ASN.2008010033
CrossRef Full Text | Google Scholar
42. Praetorius H, Frokiaer J, Nielsen S, Spring K. Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J Membr Biol (2003) 191(3):193–200. doi:10.1007/s00232-002-1055-z
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Goetz S, Anderson K. The primary cilium: a signalling centre during vertebrate development. Nat Rev Gen (2010) 11(5):331–44. doi:10.1038/nrg2774
CrossRef Full Text | Google Scholar
44. Nauli S, Alenghat F, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet (2003) 33(2):129–37. doi:10.1038/ng1076
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, et al. Loss of oriented cell division does not initiate cyst formation. J Am Soc Nephrol (2010) 21(2):295–302. doi:10.1681/ASN.2009060603
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Kunimoto K, Bayly RD, Vladar EK, Vonderfecht T, Gallagher AR, Axelrod JD. Disruption of core planar cell polarity signaling regulates renal tubule morphogenesis but is not cystogenic. Curr Biol (2017) 27(20):3120–31. doi:10.1016/j.cub.2017.09.011
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Retailleau K, Duprat F. Polycystins and partners: proposed role in mechanosensitivity. J Physiol (2014) 592(12):2453–71. doi:10.1113/jphysiol.2014.271346
CrossRef Full Text | Google Scholar
49. Ma M, Gallagher A, Somlo S. Ciliary mechanisms of cyst formation in polycystic kidney disease. Cold Spring Harb Perspect Biol (2017) 9:a028209. doi:10.1101/cshperspect.a028209
CrossRef Full Text | Google Scholar
50. Li Q, Montalbetti N, Wu Y, Ramos A, Raychowdhurry M, Cheng X, et al. Polycystin-2 cation channel function is under the control of microtubular structures in primary cilia of renal epithelial cells. J Biol Cem (2006) 281(49):37566–75. doi:10.1074/jbc.M603643200
PubMed Abstract | CrossRef Full Text | Google Scholar
51. Köttgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol (2008) 182(3):437–47. doi:10.1083/jcb.200805124
PubMed Abstract | CrossRef Full Text | Google Scholar
52. Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Beyer T, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol (2010) 12(11):1115–22. doi:10.1038/ncb2117
PubMed Abstract | CrossRef Full Text | Google Scholar
53. Chauvet V, Tian X, Husson H, Grimm D, Wang T, Hiesberger T, et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest (2004) 114(10):1433–43. doi:10.1172/JCI21753
PubMed Abstract | CrossRef Full Text | Google Scholar
54. Delling M, Indzhykulian A, Liu X, Li Y, Xie T, Corey D, et al. Primary cilia are not calcium-responsive mechanosensors. Nature (2016) 531(7596):656–60. doi:10.1038/nature17426
CrossRef Full Text | Google Scholar
55. Hofherr A, Köttgen M. Polycystic kidney disease: cilia and mechanosensation revisited. Nat Rev Nephrol (2016) 12(6):318–9. doi:10.1038/nrneph.2016.61
CrossRef Full Text | Google Scholar
57. Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss cilia suppresses cyst growth in genetic models of autosomal polycystic kidney disease. Nat Genet (2013) 45(9):1004–12. doi:10.1038/ng.2715
CrossRef Full Text | Google Scholar
58. Antignac C, Calvet JP, Germino GG, Grantham JJ, Guay-Woodford LM, Harris PC, et al. The future of polycystic kidney disease research – as seen by the 12 Kaplan awardees. J Am Soc Nephrol (2015) 26(9):2081–95. doi:10.1681/ASN.2014121192
CrossRef Full Text | Google Scholar
59. Harris PC, Hopp K. The mutation, a key determinant of phenotype in ADPKD. J Am Soc Nephrol (2013) 24(6):868–70. doi:10.1681/ASN.2013040417
CrossRef Full Text | Google Scholar
60. Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol (2013) 24(6):1006–13. doi:10.1681/ASN.2012070650
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int (2009) 75(8):848–55. doi:10.1038/ki.2008.686
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest (2012) 122(11):4257–73. doi:10.1172/JCI64313
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Fedeles SV, Tian X, Gallagher AR, Mitobe M, Nishio S, Lee SH, et al. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet (2011) 43(7):639–47. doi:10.1038/ng.860
PubMed Abstract | CrossRef Full Text | Google Scholar
66. Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med (2007) 13(12):1490–5. doi:10.1038/nm1675
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Wei W, Hackmann K, Xu H, Germino G, Qian F. Characterization of cis-autoproteolysis of polycystin-1, the product of human polycystin kidney disease 1 gene. J Biol Chem (2007) 282(30):21729–37. doi:10.1074/jbc.M703218200
CrossRef Full Text | Google Scholar
68. Yu S, Hackmann K, Gao J, He X, Piontek K, Garcia-Gonzalez MA, et al. Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc Nat1 Acad Sci U S A (2007) 104(47):18688–93. doi:10.1073/pnas.0708217104
CrossRef Full Text | Google Scholar
69. Gainullin VG, Hopp K, Ward CJ, Hommerding CJ, Harris PC. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J Clin Invest (2015) 125(2):607–20. doi:10.1172/JCI76972
PubMed Abstract | CrossRef Full Text | Google Scholar
70. Kim H, Xu H, Yao Q, Li W, Huang Q, Outeda P, et al. Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nat Commun (2014) 5:5482. doi:10.1038/ncomms6482
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Cai Y, Fedeles SV, Dong K, Anyatonwu G, Onoe T, Mitobe M, et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J Clin Invest (2014) 124(12):5129–44. doi:10.1172/JCI67273
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Guay-Woodford L. Autosomal recessive polycystic kidney disease: the prototype of the hepato-renal fibrocystic diseases. J Pediatr Genet (2014) 3(2):89–101. doi:10.3233/PGE-14092
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem (2005) 280(50):41298–306. doi:10.1074/jbc.M510082200
PubMed Abstract | CrossRef Full Text | Google Scholar
75. Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci U S A (2007) 104(15):6454–9. doi:10.1073/pnas.0610324104
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis (2010) 17(2):118–30. doi:10.1053/j.ackd.2010.01.002
CrossRef Full Text | Google Scholar
77. Hooper KM, Boletta A, Germino GG, Hu Q, Ziegelstein RC, Sutters M. Expression of polycystin-1 enhances endoplasmic reticulum calcium uptake and decreases capacitative calcium entry in ATP-stimulated MDCK cells. Am J Physiol Renal Physiol (2005) 289(3):F521–30. doi:10.1152/ajprenal.00355.2004
PubMed Abstract | CrossRef Full Text | Google Scholar
78. Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int (2000) 57(4):1460–71. doi:10.1046/j.1523-1755.2000.00991.x
PubMed Abstract | CrossRef Full Text | Google Scholar
79. Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, et al. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int (2004) 66(3):964–73. doi:10.1111/j.1523-1755.2004.00843.x
PubMed Abstract | CrossRef Full Text | Google Scholar
80. Sussman CR, Ward CJ, Leightner AC, Smith JL, Agarwal R, Harris PC, et al. Phosphodiesterase 1A modulates cystogenesis in zebrafish. J Am Soc Nephrol (2014) 25(10):2222–30. doi:10.1681/ASN.2013040421
PubMed Abstract | CrossRef Full Text | Google Scholar
81. Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med (2003) 9(10):1323–6. doi:10.1038/nm935
PubMed Abstract | CrossRef Full Text | Google Scholar
82. Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med (2004) 10(4):363–4. doi:10.1038/nm1004
PubMed Abstract | CrossRef Full Text | Google Scholar
83. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med (2012) 367(25):2407–18. doi:10.1056/NEJMoa1205511
PubMed Abstract | CrossRef Full Text | Google Scholar
85. Ibraghimov-Beskrovnaya O, Natoli TA. mTOR signaling in polycystic kidney disease. Trends Mol Med (2011) 17(11):625–33. doi:10.1016/j.molmed.2011.06.003
CrossRef Full Text | Google Scholar
86. Tao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol (2005) 16(1):46–51. doi:10.1681/ASN.2004080660
PubMed Abstract | CrossRef Full Text | Google Scholar
87. Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A (2006) 103(14):5466–71. doi:10.1073/pnas.0509694103
PubMed Abstract | CrossRef Full Text | Google Scholar
88. Renken C, Fischer DC, Kundt G, Gretz N, Haffner D. Inhibition of mTOR with sirolimus does not attenuate progression of liver and kidney disease in PCK rats. Nephrol Dial Transplant (2011) 26(1):92–100. doi:10.1093/ndt/gfq384
CrossRef Full Text | Google Scholar
89. Hogan MC, Masyuk TV, Page L, Holmes DR III, Li X, Bergstralh EJ, et al. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant (2012) 27(9):3532–9. doi:10.1093/ndt/gfs152
PubMed Abstract | CrossRef Full Text | Google Scholar
90. Chrispijn M, Nevens F, Gevers TJ, Vanslembrouck R, van Oijen MG, Coudyzer W, et al. The long-term outcome of patients with polycystic liver disease treated with lanreotide. Aliment Pharmacol Ther (2012) 35(2):266–74. doi:10.1111/j.1365-2036.2011.04923.x
PubMed Abstract | CrossRef Full Text | Google Scholar
91. Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med (2013) 19(4):488–93. doi:10.1038/nm.3092
PubMed Abstract | CrossRef Full Text | Google Scholar
92. Chiaravalli M, Rowe I, Mannella V, Quilici G, Canu T, Bianchi V, et al. 2-Deoxy-d-glucose ameliorates PKD progression. J Am Soc Nephrol (2016) 27(7):1958–69. doi:10.1681/ASN.2015030231
PubMed Abstract | CrossRef Full Text | Google Scholar
93. Zhou X, Fan LX, Sweeney WE Jr, Denu JM, Avner ED, Li X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J Clin Invest (2013) 123(7):3084–98. doi:10.1172/JCI64401
PubMed Abstract | CrossRef Full Text | Google Scholar
94. Hajarnis S, Lakhia R, Yheskel M, Williams D, Sorourian M, Liu X, et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun (2017) 8:14395. doi:10.1038/ncomms14395
PubMed Abstract | CrossRef Full Text | Google Scholar
95. Chen L, Zhou X, Fan LX, Yao Y, Swenson-Fields KI, Gadjeva M, et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest (2015) 125(6):2399–412. doi:10.1172/JCI80467
PubMed Abstract | CrossRef Full Text | Google Scholar
96. Sweeney WE, Frost P, Avner ED. Tesevatinib ameliorates progression of polycystic kidney disease in rodent models of autosomal recessive polycystic kidney disease. World J Nephrol (2017) 6(4):188–200. doi:10.5527/wjn.v6.i4.188
PubMed Abstract | CrossRef Full Text | Google Scholar
What Is the Biological Definition of a Autosomal Dominant Disorder
Source: https://www.frontiersin.org/articles/316195